
- Integral Medicine | Alexander Smithers M.D., A.P.1014 N. East Ave.
Sarasota, Fl. 34237941-444-6336 - 1014 N. East Ave.
Sarasota, Fl. 34237941-444-6336 - Testimonials
Dr Smithers is so acute and atuned to what others miss. I had been suffering with abdominal issues for over 20 years getting no help from traditional medicine. He found and correctly treated the issue. Words cannot describe the quality of life that has been restored to me. He’s very caring and takes the time to listen.
My husband had severe sciatic pain so bad he was bed ridden. Because our regular Dr. was out of town we went to our chiropractor to try to get some relief. He was not able to get as much relief a he needed so Dr. Daly asked us to see Dr. Smithers – another Dr. in his office.. What a BLESSING!! Dr. Smithers is the best of both worlds Eastern and Western medicine! He suggested something called PROLOZONE therapy... Read more »Dr. Smithers has been taking care of my knees for a few years now. My experience with him has been consistently outstanding. His care has allowed me to avoid knee replacement surgery. There was a point when I had so much pain that I was not able to spend more than 15 minutes on my feet.. Because of his treatments and direction I have gotten my life back, including 20 mile bike rides and trips to the Disney parks with
... Read more » - Testimonials
Hi
... Read more »
I had Prolozone therapy with Dr Alex Smithers. My knees and thumbs were injected due to meniscus issues in my knees and arthritis in my thumbs. Dr Alex was wonderful. He’s very gentle, kind and patient. I’m a very nervous patient and he took his time with me and did his best to make sure I was comfortable. I’m feeling better and continue to expect more and more improvement. I highly recommend Dr Alex and his staff. TheyMy husband had severe sciatic pain so bad he was bed ridden. Because our regular Dr. was out of town we went to our chiropractor to try to get some relief. He was not able to get as much relief a he needed so Dr. Daly asked us to see Dr. Smithers – another Dr. in his office.. What a BLESSING!! Dr. Smithers is the best of both worlds Eastern and Western medicine! He suggested something called PROLOZONE therapy... Read more »In Oct of 2013 I was diagnosed with stage 4 Cholangiocarcinoma Malignant Neoplasm Cancer of the Bile Ducts of the Liver. In addition I had a tumor on the left lobe of the liver that ruptured and allowed cancer cells to escape and cake on the majority of my abdominal area. The issues, pain and effects are too numerous to mention. After 9 months of Chemo Therapy Treatments I was left with major issues with neuropathy in both my feet
... Read more »I had my second appointment with Dr. Alex Smithers today. I was treated for painful arthritic hip, back, and neck. After treatments I felt immediate results. I was able to go shopping with my daughter with no pain which wouldn’t have been possible before without considerable pain. I am very thankful to my daughter who found Dr. Alex and this office. I am also very thankful to Dr. Alex. I have flown twice from Pa. To see him and will
... Read more »Thank you Dr. Alex Smithers! After dealing with lower back pain from an injury 30 years ago, I feel that after several injections, I am pain and worry free and can finally let this go.
... Read more »
I was back today for a torn knee ligament and the first ozone injection, and am confident this will be healed also.
Alex is unlike others in the medical field. He has a calm professional manner and very patient in listening and explainingDoctor Alex has been treating me for severe gut issues. I was tired of going to doctors that didn’t listen to me and would just prescribe a pill not ever trying to get to the real issues I was having. Dr. Alex has listened and treated me holistically and I am feeling great!! Thank you for everything Dr. Alex.
Uncategorized
Low-Carbohydrate Diet Superior to Antipsychotic Medications
A Cutting-Edge Conference
This summer, I was fortunate to participate in the groundbreakingInternational Society for Nutritional Psychiatry Research(ISNPR) conference held in Bethesda, Maryland. The meeting was truly inspiring and exciting to those of us who believe that nutritional approaches are the way forward in the treatment of mental healthdisorders. While the majority of the presentations at this conference were focused on omega-3 fatty acids, microbiome research, micronutrients, and the Mediterranean diet, there were a few small breakout sessions exploring the potential benefits of ketogenic diets. Ketogenic diets are special low-carbohydrate diets that have been used to treat epilepsy for almost 100 years and show great promise in the management of a wide variety of other brain disorders.
Psychosis, Mood, and Diet
One of the presentations I attended was by Dr. Chris Palmer, a psychiatrist from Harvard’s McLean Hospital in Belmont, Massachusetts. In a small room packed with curious doctors, scientists and nutritionists from around the world, Dr. Palmer described the experiences of two adults in his practice with schizoaffective disorder who had tried a ketogenic diet. Whereas schizophrenia is characterized primarily by psychotic symptoms, people with schizoaffective disorder have to cope not only with psychosis but also with overlapping periods of severe mood symptoms. Signs of psychosis include paranoia, auditory hallucinations, visual hallucinations, intrusive thoughts/images, and/or disorganized thinking. Mood episodes may include depression, euphoria, irritability, rage, suicidal thoughts, and/or mood swings. As a practicing psychiatrist for more than 15 years, I can tell you that schizoaffective disorder is a particularly challenging diagnosis for people to live with and for psychiatrists to treat. Even the most potent antipsychotic and mood stabilizing medications available often don’t bring sufficient relief, and those medications come with a significant risk of side effects.
Below I’ve summarized the cases Dr. Palmer presented. More details of each story, along with Dr. Palmer’s commentary are published in the journal Schizophrenia Research.
Case Number One: A Woman Finds Natural Relief
The first story is of a 31-year-old woman who was diagnosed with schizoaffective disorder eight years ago. Trials of TWELVE different medications, including Clozapine, a powerful antipsychotic agent considered by many psychiatrists to be the medication of last resort due to its risk of serious side effects, were unsatisfactory. She had also undergone 23 rounds of electroconvulsive therapy (ECT or what used to be called “electric shock treatments”), yet remained troubled by serious symptoms. She decided to try a ketogenic diet with the hope of losing some weight. After four weeks on the diet, her delusions had resolved and she’d lost ten pounds. At four months’ time, she’d lost 30 pounds and her score on a clinical questionnaire called the PANSS (Positive and Negative Symptom Scale), which ranks symptoms on a scale from 30 (best) to 210 (worst), had come down from 107 to 70.
Case Number Two: A Man Comes to Life
The second story is of a 33-year-old single man diagnosed with schizoaffective disorder fourteen years ago. Over the years he had tried SEVENTEEN different psychiatricmedications with limited success, including Clozapine. Weighing 322 pounds, he decided to embark on a ketogenic diet for weight loss.
Within three weeks, he reported “dramatic” reduction in auditory hallucinations and delusions, as well as better mood, energy, and concentration. Over the course of a year, he lost a total of 104 pounds. When in ketosis, his PANSS scores improved significantly—falling from 98 to only 49. His daily function and quality of life also improved dramatically; he moved out of his father’s home, began dating, and started taking college courses.
Interestingly, in both cases, each time either of these individuals went off of the ketogenic diet, their symptoms worsened, and when they went back on the diet, their symptoms improved again, suggesting it was the diet and not some other factor that was responsible.
Food vs. Medication
These outcomes are truly remarkable: improvement by dozens of points on the PANSS, significant weight loss, and better quality of life. There simply is no psychiatric medication available with the power to accomplish those results. I have certainly seen antipsychotic medications help people with bipolar and psychotic symptoms, and sometimes help dramatically. However, all antipsychotic medications, unfortunately, come with a substantial risk of side effects that can worsen quality of life, not the least of which is weight GAIN.
All antipsychotic medications (Abilify, Zyprexa, Risperdal, Seroquel, Clozapine, etc.) can contribute to high insulin levels and insulin resistance —a hormonal shift in metabolism that makes it harder for the body to process carbohydrates. Over time, insulin resistance can lead to weight gain, type 2 diabetes, heart disease, and even Alzheimer’s disease. In stark contrast, ketogenic diets have many positive side effects; they lower insulin levels and improve insulin sensitivity, reversing signs of insulin resistance and associated conditions.
What is the Ketogenic Diet?
A ketogenic diet is an ultra-low-carbohydrate diet (maximum 20 grams of carbohydrate per day) that is typically much higher in fat than other diets. This diet is designed to lower and stabilize insulin levels, allowing the body to burn fat more easily, and rely less on glucose (blood sugar) for energy. Fat is broken down into ketones, which most cells in the brain can use for energy instead of glucose. Ketones burn more cleanly and efficiently than glucose, resulting in less inflammation and oxidation throughout the brain and body.

There are many theories as to why ketogenic diets seem to be so healing and stabilizing for brain cells, some of which you can read about in this article about bipolar disorder and ketogenic diets. I have studied, written about and personally followed a ketogenic diet for the better part of the past five years, and recommend it to my patients as an alternative and/or add-on option to medication. At the ISNPR conference, I presented a poster summarizing exciting nutritional approaches to Alzheimer’s disease prevention and treatment, including ketogenic diets.
Ketogenic Diets and Other Psychiatric Disorders
Earlier this summer, I wrote an article for Psychology Today entitled Ketogenic Diets for Psychiatric Disorders summarizing studies and case reports of how low-carbohydrate and ketogenic diets affect people with psychiatric disorders including bipolar disorder, autism, and schizophrenia. That review includes the remarkable account of a woman who had suffered with psychotic symptoms for 63 years before finally experiencing relief on a low-carbohydrate diet.
Although we only have a handful of published examples so far, the information within them is full of promise for people who have been suffering from life-altering psychiatric disorders and health-compromising medications.
Hope Beyond Medication
Most people don’t realize that options beyond medication exist. It is critical that we spread awareness of these potentially powerful dietary strategies to everyone who may benefit. If you know of someone who is coping with mental illness, please share these inspiring stories with them.
If you yourself are struggling with symptoms of a mood or thought disorder, I encourage you to learn more about ketogenic diets and other nutritional approaches. Yes, medications can play a very important role in your care, but I believe that the most powerful way to change your brain chemistry is through food—because that’s where brain chemicals come from in the first place! Feeding your brain properly has the potential to get to the actual root of the problem, which may allow you to reduce the amount of medication you need to feel well and function at your best. In some cases, a ketogenic diet can even completely replace medications.
Nutritional psychiatry can empower you to take more control of your symptoms, your overall health, and the course of your future.
*Note: low-carbohydrate diets cause significant changes in body chemistry very quickly. If you take any medications or have any health problems, please do not start this diet without first consulting with your health care provider, as medication dosages may need to be closely monitored as you transition to a new way of eating. Please see this short post about the safety of ketogenic diets for more information.

-
 SHARE
SHARE -
 TWEET
TWEET -
 EMAIL
EMAIL -
 MORE
MORE
About the Author

Georgia Ede, MD, is a Harvard-trained psychiatrist and nutrition consultant practicing at Smith College. She writes about food and health on her website DiagnosisDiet.com.
Probiotic Reduces Depression and Alters Brain Activity in IBS
Gastroenterology
Happy bacteria?
Irritable bowel syndrome (IBS) has a strong mind–gut connection. Depression and anxiety often correlate with the severity of symptoms. But can the microbiome influence depression and anxiety through the reverse gut–brain connection?
This small pilot study randomized 44 patients with IBS to receive either Bifidobacterium longum or placebo for 6 weeks. Depression and anxiety scores were followed at 0, 6, and 10 weeks, and functional MRI was performed at 6 weeks.
Depression scores (but not anxiety) improved, and functional MRI showed a down regulation of fear in the amygdala and frontolimbic areas of the brain, which correlated with the improvement in depression scores.
How might bacteria influence mood?
There were no significant differences in taxonomy of the microbiome in either group, and there was no difference in inflammatory markers. But there was a change in catecholamine metabolites seen in stool samples. The authors suggest that the bacteria may influence dopamine and norepinephrine as a pathway to improve mood through the central nervous system. Of concern were the low depression scores in the placebo group at baseline. The improvement in depression scores in the B. longum group could have been due to the regression to the mean.
Here is what we believe we know
Previous research suggests the Bifidobacterium species (more than Lactobacillus species) have a beneficial influence on IBS symptoms.1,2 Eating a high-fiber, low-fat diet promotes the growth of Bifidobacterium species.3 Mind–gut therapies like gut-directed hypnotherapy work really well,4 and even open-label placebos work on IBS!5
As with all diseases, therapy is complicated and requires that we adapt to the unique ecosystem that presents with each clinical encounter. Research like this reminds us of this complexity and that there really are no clear separations between gut and mind. It’s all interconnected.
References
- Camilleri M. Probiotics and irritable bowel syndrome: rationale, putative mechanisms, and evidence of clinical efficacy. J Clin Gastroenterol. 2006;40(3):264-269. http://journals.lww.com/jcge/Abstract/2006/03000/Probiotics_and_Irritable_Bowel_Syndrome_.20.aspx
- Ford AC, Quigley EM, Lacy BE, et al. Efficacy of Prebiotics, Probiotics, and Synbiotics in Irritable Bowel Syndrome and Chronic Idiopathic Constipation: Systematic Review and Meta-analysis. Am J Gastroenterol. 2014;109(10):1547-1561. https://www.nature.com/ajg/journal/v109/n10/full/ajg2014202a.html
- Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. http://science.sciencemag.org/content/341/6150/1241214
- Lindfors P, Unge P, Arvidsson P, et al. Effects of gut-directed hypnotherapy on IBS in different clinical settings-results from two randomized, controlled trials. Am J Gastroenterol. 2012;107(2):276-285. https://www.nature.com/ajg/journal/v107/n2/full/ajg2011340a.html
- Kaptchuk TJ, Friedlander E, Kelley JM, et al. Placebos without deception: a randomized controlled trial in irritable bowel syndrome. PLoS One. 2010;5(12):e15591. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0015591
Abstract
This abstract is available on the publisher’s site.
BACKGROUND & AIMS
Probiotics can reduce symptoms of irritable bowel syndrome (IBS), but little is known about their effects on psychiatric comorbidities. We performed a prospective study to evaluate the effects of Bifidobacterium longum NCC3001 (BL) on anxiety and depression in patients with IBS.
METHODS
We performed a randomized, double-blind, placebo-controlled study of 44 adults with IBS and diarrhea or a mixed-stool pattern (based on Rome III criteria) and mild to moderate anxiety and/or depression (based on the Hospital Anxiety and Depression scale) at McMaster University in Canada, from March 2011 to May 2014. At the screening visit, clinical history and symptoms were assessed and blood samples were collected. Patients were then randomly assigned to groups and given daily BL (n=22) or placebo (n=22) for 6 weeks. At week 0, 6 and 10, we determined patients’ levels of anxiety and depression, IBS symptoms, quality of life, and somatization using validated questionnaires. At week 0 and 6, stool, urine and blood samples were collected, and functional magnetic resonance imaging (fMRI) test was performed. We assessed brain activation patterns, fecal microbiota, urine metabolome profiles, serum markers of inflammation, neurotransmitters and neurotrophin levels.
RESULTS
At week 6, 14/22 patients in the BL group had reduction in depression scores of 2 points or more on the Hospital Anxiety and Depression scale, vs 7/22 patients in the placebo group (P=.04). BL had no significant effect on anxiety or IBS symptoms. Patients in the BL group had a mean increase in quality of life score compared with the placebo group. The fMRI analysis showed that BL reduced responses to negative emotional stimuli in multiple brain areas, including amygdala and fronto-limbic regions, compared with placebo. The groups had similar fecal microbiota profiles, serum markers of inflammation, and levels of neurotrophins and neurotransmitters, but the BL group had reduced urine levels of methylamines and aromatic amino acids metabolites. At week 10, depression scores were reduced in patients given BL vs placebo.
CONCLUSION
In a placebo-controlled trial, we found that the probiotic BL reduces depression but not anxiety scores and increases quality of life in patients with IBS. These improvements were associated with changes in brain activation patterns that indicate that this probiotic reduces limbic reactivity.
Benefits of Omega-3 Fatty Acids From Fish Oil After Acute MI (Heart Attack)
What is the deal with omega-3 supplements?
Omega-3 fatty acid has been shown to reduce death post-MI in some studies; but, yet, in other studies, it has no effect. So does it work or not?
In this study, instead of looking at hard endpoints like death or MI, the researchers used cardiac MRI to look for subtle changes in the myocardium. It was a small study of 358 post-MI patients. Half of the participants received 4 gm of omega-3 supplements, and, after 6 months, these patients had less non–infarct related myocardial fibrosis. They also had a reduced left ventricular systolic volume index by 5.8%. They had lower inflammatory markers in their blood, and the higher the levels of omega-3, the better the results; so, the effect seen with omega-3 seemed to be dose-dependent.
Now, before we go betting the farm on omega-3 supplements, let’s go back to some basics of what omega-3 fatty acids really are.
Fatty acids are long chains of carbons with hydrogens attached to them. The tail is referred to as the omega end. These fatty acids are named by where the first double bond is between the carbons. So, omega-3 means that the first double bond is at the third carbon counted from the tail end, the omega end. Omega-6 is a fatty acid that has the first double bond at the sixth carbon.
Both omega-3 and -6 are essential fatty acids, meaning that our bodies cannot make them so we need to eat them in our diet. Many chemicals needed by the body are made from both omega-3 and -6. However, one family of chemicals that can be produced from them is called eicosanoids. This family includes some very familiar chemicals like leukotrienes (inflammation), thromboxane (blood clots), and prostaglandins, which cause inflammation and other effects in the body.
Omega-6 is processed in the body much faster than omega-3, and so it creates more “bad” chemicals. A lot of omega-6 produces a lot of bad chemicals, but more omega-3 counterbalances this, which is why we think of omega-3 as good. In reality, however, omega-3 is simply not as bad as omega-6. Experts believe that we should be eating at a 1:1 ratio of omega-6 to omega-3.
Unfortunately, our western diet is more like 10:1 to 30:1 of omega-6 to omega-3. Too much omega-6 means we make a lot of inflammatory and thrombotic chemicals. Our diet has evolved to higher omega-6 content over the years. For example, corn oil is 46:1 omega-6 to omega-3. The ratio in grass-fed cows is 2:1, but it is more like 4:1 in grain-fed cows. There is less omega-3 in grains than in grass, and so grain-fed cows have less omega-3.
Perhaps to make sense of these studies we need to look at the omega-6 to omega-3 ratios. In the old studies, 1 gm of omega-3 was given to the participants, but if the participants had ratios of 30:1 then that 1 gm would not have had any impact. So, perhaps using the ratio to separate out the patients may give us more consistent outcomes.
For now, how should we improve the ratio? Omega-3 is made in plants, seaweed, and algae. So we can eat more plant-based foods and seaweed. Fish eat the algae, which is how they become rich in omega-3, so we can eat fish. Some estimates put omega-3 at seven times the level of omega-6 in fish; but, remember, that the larger the fish, the greater the amounts of heavy metals they accumulate. So, we should not eat too much of the big fish like tuna but perhaps should focus more on the small, oily fish, which have not lived long enough to accumulate these toxins. For meat, we should try for that from grass-fed livestock.
Perhaps the key is to balance our omega-3 and -6. Maybe it’s simpler to just take a supplement as was done by participants in this study; but, for the majority of us, perhaps we should choose foods that have a better ratio of omega-3 to omega-6. Remember, we can’t make these so our bodies will have to use what we eat. So choose wisely.
Abstract
This abstract is available on the publisher’s site.
BACKGROUND
Omega-3 fatty acids from fish oil have been associated with beneficial cardiovascular effects, but their role in modifying cardiac structures and tissue characteristics in patients who have had an acute myocardial infarction while receiving current guideline-based therapy remains unknown.
METHODS
In a multicenter, double-blind, placebo-controlled trial, participants presenting with an acute myocardial infarction were randomly assigned 1:1 to 6 months of high-dose omega-3 fatty acids (n=180) or placebo (n=178). Cardiac magnetic resonance imaging was used to assess cardiac structure and tissue characteristics at baseline and after study therapy. The primary study endpoint was change in left ventricular systolic volume index. Secondary endpoints included change in noninfarct myocardial fibrosis, left ventricular ejection fraction, and infarct size.
RESULTS
By intention-to-treat analysis, patients randomly assigned to omega-3 fatty acids experienced a significant reduction of left ventricular systolic volume index (–5.8%, P=0.017), and noninfarct myocardial fibrosis (–5.6%, P=0.026) in comparison with placebo. Per-protocol analysis revealed that those patients who achieved the highest quartile increase in red blood cell omega-3 index experienced a 13% reduction in left ventricular systolic volume index in comparison with the lowest quartile. In addition, patients in the omega-3 fatty acid arm underwent significant reductions in serum biomarkers of systemic and vascular inflammation and myocardial fibrosis. There were no adverse events associated with highdose omega-3 fatty acid therapy.
CONCLUSIONS
Treatment of patients with acute myocardial infarction with high-dose omega-3 fatty acids was associated with reduction of adverse left ventricular remodeling, noninfarct myocardial fibrosis, and serum biomarkers of systemic inflammation beyond current guidelinebased standard of care.
Prebiotic Reduces Body Fat and Alters Intestinal Microbiota in Overweight or Obese Children
- Written by
This small study of overweight children found that taking a prebiotic (oligofructose-enriched inulin, 8 g daily for 16 weeks) was associated with less weight gain than placebo and resulted in a shift in the microbiome to include more bifidobacteria. Bifidobacteria is established with breastfeeding and is associated with less weight gain (see graphic 1 for the results of the study).

But, before you Google “oligofructose-enriched inulin” for a supplement to recommend to your overweight patients, remember that this is simply a fiber combined with short-chain sugars found in plants. A prebiotic is generally a soluble fiber that bypasses absorption in the upper intestinal track, passing into the colon and providing fuel for bacterial growth and fermentation (gas; see graphic 2).

Foods rich in inulin include whole grains, onions, garlic, leeks, Jerusalem artichoke, and chicory root. Other prebiotic fibers that would likely work include psyllium, ground flax seed, and guar gum. The good news is that, other than gas and bloating, these products are very safe and may promote the establishment of a healthy microbiome at an early age, which may help with weight management as the child grows into an adult.
Abstract
This article is open access.
BACKGROUND & AIMS
It might be possible to manipulate the intestinal microbiota with prebiotics or other agents to prevent or treat obesity. However, little is known about the ability of prebiotics to specifically modify gut microbiota in children with overweight/obesity or reduce body weight. We performed a randomized controlled trial to study the effects of prebiotics on body composition, markers of inflammation, bile acids in fecal samples, and composition of the intestinal microbiota in children with overweight or obesity.
METHODS
We performed a single-center, double-blind, placebo-controlled, trial of 2 separate cohorts (March 2014 and August 2014) at the University of Calgary in Canada. Participants included children, 7 – 12 years old, with overweight or obesity (>85th percentile of body mass index) but otherwise healthy. Participants were randomly assigned to groups given either oligofructose-enriched inulin (OI, 8 g/day; n=22) or maltodextrin placebo (isocaloric dose, controls; n=20) once daily for 16 weeks. Fat mass and lean mass were measured using dual-energy-x-ray absorptiometry. Height, weight, and waist circumference were measured at baseline and every 4 weeks thereafter. Blood samples were collected at baseline and 16 weeks, and analyzed for lipids, cytokines, lipopolysaccharide, and insulin. Fecal samples were collected at baseline and 16 weeks; bile acids were profiled using high-performance liquid chromatography and the composition of the microbiota was analyzed by 16S rRNA sequencing and quantitative PCR. The primary outcome was change in percent body fat from baseline to 16 weeks.
RESULTS
After 16 weeks, children who consumed OI had significant decreases in body weight z-score (decrease of 3.1%), percent body fat (decrease of 2.4%), and percent trunk fat (decrease of 3.8%) compared to children given placebo (increase of 0.5%, increase of 0.05%, and decrease of 0.3%, respectively). Children who consumed OI also had a significant reduction in level of interleukin 6 (IL6) from baseline (decrease of 15%) compared with the placebo group (increase in 25%). There was a significant decrease in serum triglycerides (decrease of 19%) in the OI group. Quantitative PCR showed a significant increase in Bifidobacterium spp. in the OI group compared with controls. 16S rRNA sequencing revealed significant increases in species of the genus Bifidobacterium and decreases in Bacteroides vulgatus within the group who consumed OI. In fecal samples, levels of primary bile acids increased in the placebo group but not in the OI group over the 16-week study period.
CONCLUSIONS
In a placebo-controlled, randomized trial, we found a prebiotic (OI) to selectively alter the intestinal microbiota and significantly reduce body weight z-score, percent body fat, percent trunk fat, and serum level of IL6 in children with overweight or obesity.
Acupuncture Found to Be an Effective Analgesia Option in ER
May provide a safe alternative to opioids, researchers say
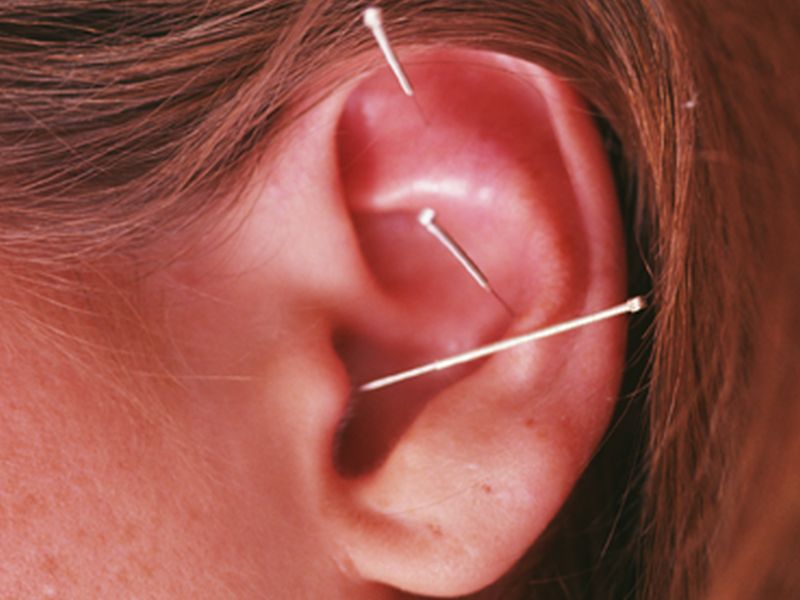
Acupuncture is a safe and effective alternative to pain medications for some emergency department patients, according to a study published in the June 19 issue of the Medical Journal of Australia.
MONDAY, June 19, 2017 (HealthDay News) — Acupuncture is a safe and effective alternative to pain medications for some emergency department patients, according to a study published in the June 19 issue of the Medical Journal of Australia.
The study — billed as the world’s largest randomized, controlled trial of acupuncture in the emergency department — included 528 patients. The study participants were seen at four Australian emergency departments for acute low back pain, migraines, or ankle sprains. Patients who said their level of pain was at least 4 on a 10-point scale received one of three treatments: acupuncture alone; acupuncture with pharmacotherapy; or pharmacotherapy alone.
One hour after treatment, 36.9 percent of all patients had significant pain reduction, meaning at least a 2-point decline on the 10-point scale. More than 80 percent still had a pain rating of at least 4, the researchers found. But two days later, most patients were satisfied. Overall, 82.8 percent of acupuncture-only patients said they would probably or definitely repeat their treatment, compared with 80.8 percent in the combined group and 78.2 percent in the pharmacotherapy-alone group.
“Emergency nurses and doctors need a variety of pain-relieving options when treating patients, given the concerns around opioids such as morphine, which carry the risk of addiction when used long-term,” lead investigator Marc Cohen, M.B.B.S., Ph.D., a professor in the School of Health and Biomedical Sciences at RMIT University in Melbourne, Australia, said in a university news release.